药物代谢研究在药物研发中的作用
设计活性高、选择性强、毒副作用小、应用广泛的新药不但是药物设计人员孜孜以求的目标,也是临床用药发展的需要。在新药设计中,很多优秀药物并不是完全新型结构的化合物,而是通过对先导化合物或老药结构进行合理改进或修饰而开发出来,它们往往具有更理想的理化性质或者药动学性质,或者膜透过性增强、吸收和生物利用度提高,或者具有靶向作用,选择性提高,毒副作用减弱。
设计活性高、选择性强、毒副作用小、应用广泛的新药不但是药物设计人员孜孜以求的目标,也是临床用药发展的需要。在新药设计中,很多优秀药物并不是完全新型结构的化合物,而是通过对先导化合物或老药结构进行合理改进或修饰而开发出来,它们往往具有更理想的理化性质或者药动学性质,或者膜透过性增强、吸收和生物利用度提高,或者具有靶向作用,选择性提高,毒副作用减弱。对药物结构进行修饰时,常常需要掌握药物代谢的规律,如药物代谢部位、代谢酶种类、代谢形式和途径、代谢产物等,药物代谢研究可以为结构修饰提供参考或依据,对于发现疗效更好、作用更强、毒副作用更低的新药物具有重要的实际意义。
1前体药物的设计
1.1 起全身作用的前体药物
前体药物的设计思想就是利用药物在体内或组织内经水解或氧化代谢,形成活性产物而发挥作用。设计前体药物一个主要目的是为了提高口服吸收百分率。例如,对于含有羧基或者(酚)羟基的药物,由于极性大,生物膜透过性较差,口服吸收率一般不高,可以利用酯化或者酰胺化的方法对其结构进行修饰,增加脂溶性,提高膜透过性和口服吸收率。抗生素类前体药物大多利用这种方法改造而成。如氨卡青霉素口服吸收率为50%,将其极性羧基酯化后得到的一个前体药物吡呋氨卡青霉素,亲脂性增加,口服吸收率高达98%-99%。又如,依那普利为常用的ACE抑制剂,为具有活性的依那普利拉的乙酯化前体药物,依那普利拉的胃肠道吸收少于10%,而依那普利的口服吸收达60%,试验证实肝脏代谢使之水解转换为活性二酸结构。
除了利用酯化和酰胺化的方法制成前体药物外,还可以用更复杂的方法设计前体药物。例如,阿昔洛韦为一种有效的抗疱疹病毒药,口服吸收差且无规律,其结构上有一个可进行衍生化的羟基,对此羟基进行酯化却不能增加吸收,但是科研人员设计出的另一个前体药物地昔洛韦可以经过黄嘌呤氧化酶氧化为具有抗病毒活性的阿昔洛韦,地昔洛韦口服吸收较阿昔洛韦或其酯均高。因此除了考虑水解代谢途径外,对于酯化和酰胺化制成的前体药物口服吸收不理想,还可以针对特定药物结构和代谢方式,设计其它代谢形式的前体药物,这需要进行深入的代谢研究。
1.2 起局部作用的前体药物
一般来说,由于非特异性酯酶在全身分布广泛,因此对于发挥全身作用的药物可以制成酯类前体药物。对于需要在特定部位起作用的药物,就需要弄清酶的分布和代谢方式,制成相应的前体药物,在特定部位酶作用下产生活性代谢物而发挥作用,而身体其它部位和器官由于缺乏或者根本没有相应的代谢酶,很少或不产生代谢产物,从而有效避免药物对这些部位产生毒副作用,提高选择性,减少用药量。
1.2.1 中枢神经系统药物
血脑屏障中毛细血管连接紧密,对于作用于中枢神经系统的药物,转运主要障碍是毛细管上皮细胞双层脂膜,药物要通过血脑屏障,需要具有一定的脂溶性。研究表明,药物进入脑脊液的速度与其在PH7.4时的油水分配系数几乎成正比。例如,分配系数高的硫喷妥、苯胺、氨基比林容易透过血脑屏障,而分配系数低的N-乙酰-4-氨基安替比林和磺胺脒的透过性极差。另外,对吩噻嗪类安定药,如氟丙嗪、异丁嗪、氯丙嗪、氟吩嗪、丙嗪的研究发现,化合物的亲脂性是药物能否透过血脑屏障的决定因素,亲脂性强,脑内药物浓度高。因此,在设计中枢神经系统药物时必须考虑其脂溶性,对于含有极性基团如羟基或者羧基的药物可以通过酯化形成前体药物以增加脂溶性或油水分配系数的方法增强其透过血脑屏障的能力,在到达中枢神经系统后,药物在大脑内含有的羧酸酯酶作用下水解,形成活性产物。例如,吗啡经3,6-二羟基乙酰化成为海洛因(二乙酰吗啡)脂溶性大大增加,其脑摄取较吗啡竟增加25倍多,当海洛因进入大脑后,很快被代谢转化为单乙酰吗啡和吗啡。
1.2.2 肾脏用药
左旋多巴是神经递质多巴胺的前体药物,除了起到神经递质的作用外,其还在肾脏中发挥由受体调节的血管舒张作用。γ-谷氨酰多巴是左旋多巴的前体药物,小鼠腹腔注射的γ-谷氨酰多巴在肾脏中一系列γ-谷氨酰转肽酶和L-芳香氨基酸脱羧酶作用下产生多巴胺;在服用γ-谷氨酰多巴后,肾脏中多巴胺浓度为口服等剂量左旋多巴的5倍。大鼠灌注10 nmol(g.30min)γ-谷氨酰多巴使肾脏血流量增加60%,而同剂量的左旋多巴不影响肾脏血流量。γ-谷氨酰多巴的选择性作用正是在酶的作用下实现的,由于酶在身体各部位的分布不同,γ-谷氨酰多巴在身体其它部位不能或者很少代谢形成多巴胺,而在肾脏中产生的多巴胺浓度高,结果该药显示出选择性的肾脏血管舒张作用,而对身体其他部位的副作用小。
2化合物结构修饰或引入合适基团
氟康唑的发现就是成功地利用了改变结构从而改变代谢的典例。辉瑞公司首先发现了一个理想的抗真菌药噻康唑,临床上局部用药时对阴道和皮肤真菌感染有效,但是静注或口服时,效果很差;药动学研究表明,该药在胃肠道吸收好,但首过代谢强,导致生物利用度低,这是因为噻康唑的脂溶性高,代谢速度快;为了减少其代谢,研究人员用1,2,4-三唑基团替代咪唑基团以降低亲脂性,得到了一个三唑化合物,几种动物口服药动学研究显示,其吸收和动力学性质良好,但是发现它对大鼠和狗肝毒性较强,考虑到这可能是因为引入2,4-二氯苯基所致;研究人员用氟原子取代分子中的氯原子,结果降低了肝毒性。成功地开发出新一代抗真菌药氟康唑。无独有偶,水杨酸的衍生物二氟尼柳就是在苯环的邻位和对位引入了氟,与原化合物相比,引入氟后的化合物在体内的代谢速度减慢,药理作用时间大为延长。
很多药物具有苯环结构,从药物代谢规律看,苯环结构容易被羟基化而失活,所以在进行药物设计时,就可以考虑在苯环易氧化的邻位或对位引入合适的基团以阻止其氧化,这些基团可以是氟/氯/三氟甲基/叔丁基等,这种引入一般结合牢固,不影响原来化合物的药理活性,甚至还可增强活性。
3药物活性代谢产物
药物在体内的代谢产物研究不但是药物代谢研究不可或缺的重要组成部分,也是新药发现的一个重要源泉。一般情况下,生物转化的过程就是药理学上活性化合物的失活过程,但是现在发现许多药物的代谢产物具有较原药更高的药理活性或者更理想的药动学性质,而被开发成高效/低副作用的药物。活性代谢产物被用于新药筛选来源的最典型例子是对乙酰氨基酚,它为非那西丁的O-去乙基代谢物,与非那西丁相比,其解热镇痛作用更强,其另一突出优点是不引发高铁血红蛋白血症和溶血性贫血;非那西丁可以通过O-去乙基和N-去乙酰基及水解形成十多种代谢物,其代谢物N-羟基乙氧基苯胺是产生上述副作用的原因;而对乙酰氨基酚只经葡萄糖醛酸化和硫酸化代谢,因此其在临床使用剂量下十分安全。同样,羟基保泰松是保泰松的活性代谢物,与对乙酰氨基酚一样,其镇痛作用更强,而胃肠道刺激性更小。
活性代谢物除了具有良好的药效且副作用少外,其药动学性质还可能较原药更好。例如,很多苯二氮卓类药物在体内形成活性代谢物,具有相似的药理性质。奥西泮是利眠宁/哈拉西泮/氯氮卓盐/地西泮的共同代谢物,与其它苯二氮卓类不同,奥西泮只在体内产生葡萄糖醛酸化反应,半衰期更短,对于治疗失眠而言,由于在发挥作用后很快从体内消除,副作用小,具有明显的药动学优势,因此成为治疗失眠短效良药。
4硬药和软药
硬药是指不被代谢的药物。硬药主要通过肾排泄,动物种属和个体之间的消除差异就主要由相应动物或个体的肾功能决定。硬药不但解决了产生中间产物和活性代谢产物而带来的毒性问题,而且因为药物仅通过胆汁或者肾排泄,其体内药动学行为也大大简化。但在药物开发中,由于具有刚性结构的活性化合物种类稀少,且体内酶代谢功能很强,使得开发成功的硬药十分有限。几种开发成功的硬药包括治疗骨质疏松的药物二磷酸盐和某些ACE抑制剂。二磷酸盐类药物在动物和人体内不被代谢,其消除的唯一途径是肾排泄,从安全性角度来看,由于不被代谢,无毒性代谢物产生,因此使用安全。
软药是指本身具有药理活性,在体内以可预料和可控方式代谢为无毒和无药理活性的代谢产物的药物,因为绝大多数氧化反应由细胞色素P450酶系统介导,而这种代谢常常受到年龄/性别/疾病/种族和环境因素的影响,从而导致同一药物在体内的生物转化和药代动力学变化存在较大的个体差异,不易控制和预测。软药的主要设计思想是尽可能避免氧化代谢,一般利用水解酶即得到可以预料的和可控的药物代谢方式。
阿曲库铵为一种非去极化肌肉松弛药,它含一个季铵基团和酯结构,其在体内通过两个非氧化途径代谢:一个为非酶代谢——霍夫曼降解,形成叔铵和烯链,另一个经酯酶水解。一般设计的酯类和酰胺类前体药物虽然在代谢形式上与软药相似,但是两者具有本质的差别,软药具有活性,经过代谢成为无毒和无活性的产物,而前体药物本身无活性,经过代谢转化为具有药理活性的产物。
5代谢毒性
许多药物能通过代谢激活形成反应性代谢产物,并可能产生药物诱导的毒性作用,而药物的分子结构与这种代谢诱导的毒性有直接的相关性。如果该结构为药物的必要活性基团,则可通生物电子等排等原理将易产生毒性的结构用弱代谢基团代替,从而达到低毒性的目的。如噻氯匹定是一种抗血小板药物,在氧化条件下噻氯匹定易被活化成噻吩-S-氯化物,并进一步反应生成可与谷胱甘肽共价结合的产物;抗炎药替尼达普是一个双环氧合酶和5-脂肪氧合酶抑制剂,代谢动力学证明,该药有较强的肝脏毒副作用。
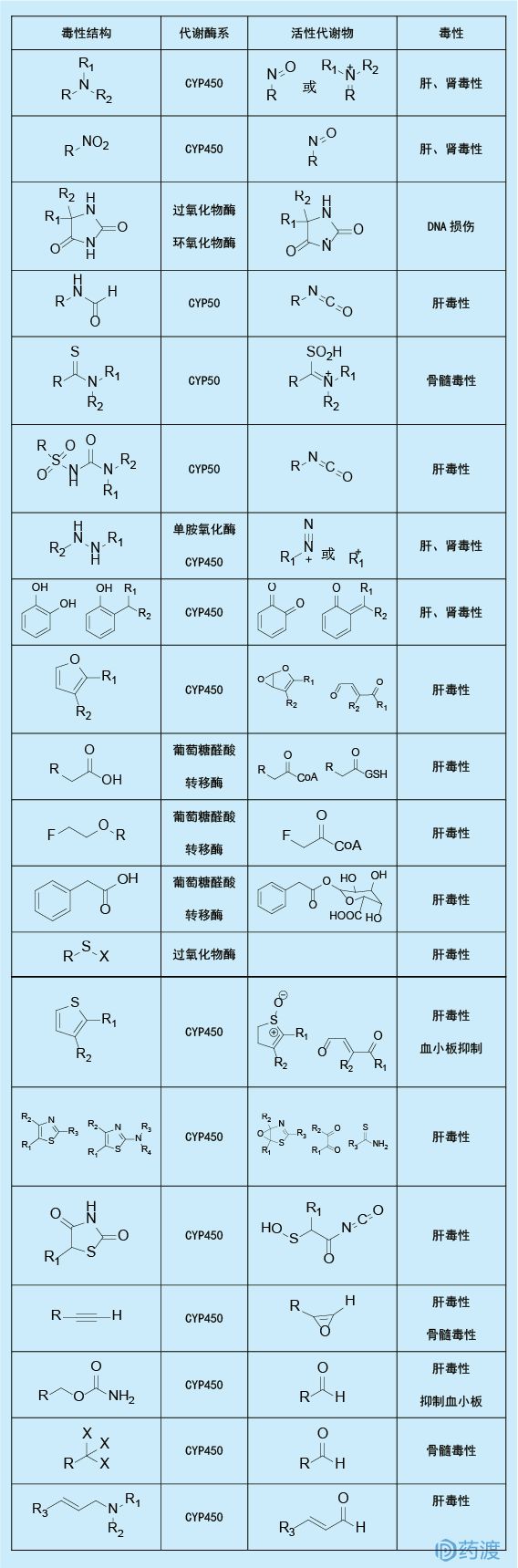
通过适当的药物代谢研究,不但可以为新药设计提供有益信息和指导,还可对设计出的药物进行合理评估。随着药物设计新技术的不断发展,药物代谢研究必将在具有高效、低毒、低副作用的靶向药物设计中起着更加重要的作用。
参考资料:
1. 生物药剂学
2. 药剂学
3. Roleof pharmacokinetics and metabolism in drug discovery and development.
相关文章
评论

 我要跟帖
我要跟帖




