肌张力障碍诊治进展:这五大改变,你都知道吗?
1911年,德国神经学家Hermann Oppenheim首先提出了“肌张力障碍”的概念,并推测一组少见运动障碍患者的核心缺陷是肌张力异常。而今,肌张力障碍已不再被认为是肌张力异常所致,因此,该如何准确定义和描述其特征已引发了多次的争论。
1911年,德国神经学家Hermann Oppenheim首先提出了“肌张力障碍”的概念,并推测一组少见运动障碍患者的核心缺陷是肌张力异常。而今,肌张力障碍已不再被认为是肌张力异常所致,因此,该如何准确定义和描述其特征已引发了多次的争论。
一种新的分类系统
最近,一个国际共识委员会正在尝试修改肌张力障碍的定义,以解决其先前定义的局限性。他们将肌张力障碍定义为:
“一种以持续或间歇性肌肉收缩所致的动作和/或姿势异常为特征的运动障碍。这种运动障碍常反复发作、有典型发作模式和肢体扭曲,并可伴有震颤。”
此外,有助于和其他运动障碍相区别的肌张力障碍独特表型也被添加到了新定义中:
“肌张力障碍常常因自主运动引发或恶化,且伴有过度的肌肉活化。”
尽管上述修改后的定义解决了先前定义的一些不足,但也引发了一些争论。例如,现在的定义认识到,肌张力障碍可以表现为不连续、不规则的肌肉收缩,而且肌张力障碍的姿势异常可能是强直或痉挛性的、动态或固定性的,或是这些表现方式的任意组合。
除提议修改了肌张力障碍的定义外,上述国际共识委员会还在着手改良肌张力障碍的分类。在1976年,Fahn和Eldridge首先提出,应将有或无遗传模式的原发性肌张力障碍与那些由已知环境因素或其他遗传性神经系统疾病所致的继发性肌张力障碍区分开来。尽管这个分类方案今天仍然有用,但对于肌张力障碍各种病因越来越多的了解,提示我们要有一个更为详细的分类系统。
肌张力障碍的不同表现促成了基于临床特征的分类方案。其中最为流行的是根据发病年龄和身体分布特征进行的分类,且常用26岁作为早期和迟发性肌张力障碍类型的年龄切点。然而,该分类方法只将DYT1型肌张力障碍作为了关注重点。随着更多肌张力障碍遗传形式的确定,以及人们对年龄和具体肌张力障碍病因相关性认识的增加,对年龄分类的细化需求目前已很明显。此外,人们对于肌张力障碍的多种时间模式,以及通过临床特征识别患者潜在诊断认识的增加,也提示我们要有更新的分类方案。
上述国际共识委员会提出了基于病因和临床特征这两大主轴而重建的肌张力障碍分类系统(表1)。
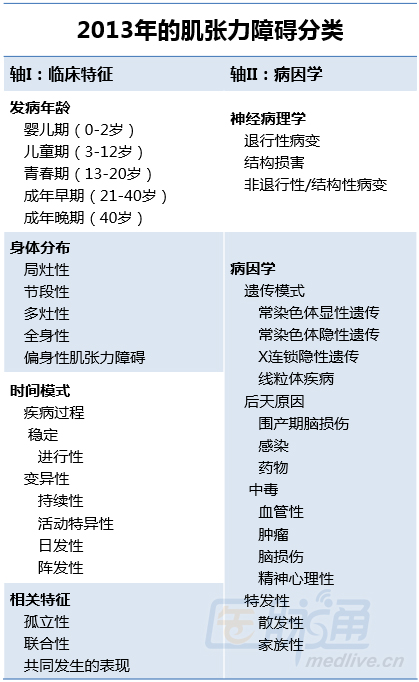
其中,临床特征轴包括了存在已久的、按发病年龄和身体分布进行的分类,但将发病年龄分为了5个年龄组,以便更好地区分那些倾向于累及这些不同年龄段个体的不同病因。修订后的分类系统还包括了一个以疾病时间模式为关注点的新分类,这将有助于肌张力障碍具体类型的诊断,并指导其治疗。肌张力障碍的时间特征被分为2类,分别用于描述其整体的疾病过程和短期内的变化。
新提出的肌张力障碍分类系统一个有争议的方面是,删除了此前描述病因时广泛使用的“原发性”和“继发性”术语。虽然这些术语在以往有助于两个主要病因分类之间的区分,但其概念不够清晰,且有效性已逐渐下降。而有效性下降的部分原因是因为人们对于多种多样的肌张力障碍病因的更多理解,及其对肌张力障碍相关神经病学和精神病学特征认识的日益增加。
因此,目前提出的方法是根据肌张力障碍的相关特征进行分类。也就是说,要么是孤立性的(除了震颤以外,仅有运动方面的特征),要么是伴有其他神经系统或全身表现。
第二大主轴旨在通过病因学对肌张力障碍的不同形式进行分类。该方法基于共同的生物学机制对众多的肌张力障碍病因进行分组。其中包括,根据是否存在可识别的解剖学变化,或是否为遗传性、后天性或特发性,来对肌张力障碍进行分组。而进一步的亚分组,可勾勒出肌张力障碍的遗传模式或获得性肌张力障碍的具体环境原因。此外,随着人们对于肌张力障碍遗传和环境促成因素的进一步理解,对一些运动障碍进行再分类,并对这一分类方案进行修订都是可能的。
非运动特征的重要意义
肌张力障碍是根据其运动表现来定义的,而且孤立性(以前的原发性)肌张力障碍长期被视为一种纯粹的运动障碍性疾病。然而,人们已越来越多地意识到其非运动特征的重要性。和帕金森病一样,肌张力障碍的非运动症状,如疼痛、睡眠障碍、精神症状、疲劳和认知功能障碍等,都可能影响患者的生活质量,并在很大程度上促成了其残疾的发生。
因此,神经学家越来越需要认识和解决肌张力障碍患者的非运动特征,因为后者会对患者的治疗满意度产生很大影响。
对于眼睑痉挛、喉部和颈部肌张力障碍患者而言,其肌张力障碍发生前常有眼部干涩、咽喉和颈部不适症状。疼痛可分别见于约75%的颈部肌张力障碍和30%的书写痉挛患者。睡眠障碍在各类局灶性肌张力障碍患者中均有报告,其中包括睡眠效率和质量下降、REM睡眠减少、觉醒增加,以及觉醒前异常运动增加等。
一项涉及221例局灶性肌张力障碍患者的新近研究显示,眼睑痉挛和颈部肌张力障碍患者,报告有睡眠质量受损的比例分别为44%和46%,而健康对照者的这一比例则为20%。该研究没有发现睡眠障碍和肌张力障碍严重程度之间存在相关性,这增加了睡眠异常可能是孤立性肌张力障碍一种内在特征的可能性。但该研究发现睡眠障碍与患者的抑郁症状相关,提示抑郁症或抗抑郁药使用,可能是上述研究结果的促成因素。
情绪障碍是肌张力障碍患者最常见的精神症状,也是患者生活质量的预测因子。最近的一项综述分析,纳入了基于规范化临床访谈和诊断标准进行的较大相关研究的89例局灶性肌张力障碍患者。其结果发现,有57.3%的受试患者存在精神疾病,而健康受试者和面肌痉挛者的这一比例则分别为24.1%和34.6%。由于相关精神疾病都是在患者的肌张力障碍发病前出现,且两者发生的平均时间间隔为18.4士13.9年,所以,不支持精神疾病是继发于肌张力障碍运动症状的想法。
一些研究还发现,抑郁症在DYT1型肌张力障碍非显性携带者中更常见。而其他类型肌张力障碍的抑郁症状与其肌张力障碍严重程度无关,进一步支持抑郁可能是肌张力障碍的本质特征,而不只是对运动障碍的一种反应。然而,另一些研究也报告了治疗相关的运动症状变化与肌张力障碍患者情绪改善之间的相关性,并使肌张力障碍患者的抑郁原因探索变得更加复杂。
虽然焦虑和抑郁在肌张力障碍患者中常常相伴发生,但焦虑(如社交恐惧症和强迫症)与肌张力障碍之间的关系仍不明确。一项涉及116例颈部肌张力障碍患者的研究显示,其社交恐惧症的终生患病率为71%。同样也有报告显示,肌阵挛性肌张力障碍综合征(DYT11)患者的强迫症发生率增加。但在其他类型的肌张力障碍患者中,还没有发现其焦虑症有更普遍的增加。
除了精神症状以外,许多研究还探讨了特发性和遗传性肌张力障碍患者的认知缺陷。尽管已有报告显示肌张力障碍患者和对照组之间,在注意力、执行力和视空间功能性方面存在一些重要差异,但这些研究结果大都偏中性,且可能通过多巴胺或抗胆碱能药物治疗暴露,或存在疼痛、睡眠障碍及抑郁等混杂因素来加以解释。总体而言,孤立性肌张力障与临床相关的认知功能障碍之间是否存在相关性,目前仍缺少足够的相关证据。
新发现的孤立性肌张力障碍基因
遗传学研究的进展已经使孤立性肌张力障碍单基因病因的认定数量快速增长(表2),并使人们对肌张力障碍的发病机制有了越来越多的了解。

1997年,有研究首先报告DYT1型肌张力障碍是由影响TorsinA(TOR1A)蛋白的基因突变所致的一种显性遗传、早发、全身性的孤立性肌张力障碍。TOR1A基因是伴侣蛋白AAA1蛋白家族(与多种细胞活动相关的ATP酶)中的一员,并与核膜、内质网和细胞骨架等多种细胞结构相关。尽管对于TOR1A基因的功能已进行了近20年研究,但这种基因突变是如何导致了肌张力障碍的发生,目前仍不清楚。
第二个孤立性肌张力障碍-DYT6,是在2009年被确定。其是一种青少年时期发病的混合性肌张力障碍,由包含细胞凋亡相关蛋白1基因(THAP1)的凋亡相关蛋白结构域的主要显性遗传基因突变所引起。THAP1是一种锌指蛋白,可参与多个靶基因的基因转录调节。
通过下一代测序和全基因组连锁分析相结合,确定可导致孤立性肌张力障碍的基因突变数量,在过去的2年中已经翻了一番多。而Cip1-相互作用锌指蛋白1基因的突变(CIZ1)(DYT23)最近已被认定为成年期发病的颈部肌张力障碍的原因。
此外,anoctamin 3基因突变(ANO3)(DYT24)和鸟嘌呤核苷酸结合蛋白(G蛋白)、α激活活性多肽、嗅觉基因(GNAL)(DYT25)也被认定为成年起病的颅颈部肌张力障碍的原因。尽管有类似的临床表型,这3种最近发现的基因却有不同的功能,包括细胞周期调控(C/27)、钙离子门控氯离子通道(ANO3)、以及多巴胺D1受体和腺苷A2a受体信号(GNAL)等。
迄今为止,已有可导致孤立性肌张力障碍的基因突变报告,使我们对肌张力障碍的分子机制有了一些深入了解,并促成了孤立性肌张力障碍分子通路话题的出现。例如,TOR7A 和GNAL突变都可干扰多巴胺信号,这与提示多巴胺功能异常在肌张力障碍发病中作用的其他证据是一致的。TOR7A、THAP7和CIZ7已被证明与细胞周期调控和转录调控有关,强化了这些关键的细胞功能缺陷可能对肌张力障碍的发病有重要作用。此外,TOR7A和THAP7编码蛋白对于核膜和内质网功能的重要性,提示其维持细胞存活作用的紊乱,可能是理解肌张力障碍发病机制的关键一环。
肌张力障碍是一种神经网络性疾病
虽然孤立性肌张力障碍没有神经退行性变的证据,但其多种细微的微观结构变化已有报道。此外,使用结构和功能成像,以及其他的生理研究工具,目前已检测到多个受累脑区的异常。考虑到不同系列的实验结果,以及人们对于基底节以外脑结构参与肌张力障碍发病认识的日益加深,许多研究者提出,将肌张力障碍视作一种神经网络性疾病,可能是对其最好的理解。
有相当多的证据表明,与感觉运动有关的基底神经节和丘脑皮质环路,可能在肌张力障碍的病理生理中发挥了关键作用。但一些成像研究已经检测到了这些环路以外的脑部异常,其中包括不同的脑皮层区域(如顶叶和扣带回皮质)、脑干和小脑等。相关病变的研究也表明,基底节、丘脑、皮层,以及脑干、小脑和脊髓病变,都可以导致肌张力障碍。此外,动物模型研究提供的额外证据也清楚地表明,基底神经节和小脑功能障碍都可能引起肌张力障碍。
在神经网络模型中,相关神经网络内的一个或更多脑区的功能障碍或其脑区之间的通讯异常,都可导致肌张力障碍发生。由于小脑和相关丘脑环路的作用得到了越来越多的认识,人们推测,肌张力障碍可能是由基底神经节和小脑网络之间不正常的相互作用所引起(见下图)。
具有肌张力障碍潜在相互作用区域的基底神经节网络(红色)和小脑网络(蓝色),包括丘脑和脑桥核
这种神经网络性疾病的概念,可能有助于解释已知孤立性肌张力障碍基因所涉及的不同细胞通路,何以都能导致肌张力障碍发生。此外,这也助于对肌张力障碍先前的大量影像学结果及动物模型和病变研究结果进行解释。
肌张力障碍的神经网络性疾病模型是一个相对较新且宽泛的概念范畴,力求能涵盖肌张力障碍发病机制方面的现有证据。这一概念增加了人们对于演变中的肌张力障碍的理解,并有助于激发新的、可检验假说的产生。这种神经网络模型是否预示着新的肌张力障碍治疗方法将被开发,及其是否能与未来的基因、神经病理学、动物模型和影像学研究结果保持兼容,仍有待观察。
新型肉毒杆菌毒素
目前常用于治疗肌张力障碍的口服药物包括抗胆碱能药物、巴氯芬、苯二氮卓类药物、多巴胺调节剂,以及其他的肌肉松弛剂。这些药物主要是对症治疗,通常只能提供有限的获益,且副作用很大。而肉毒杆菌毒素(BoNT)已被证明是许多肌张力障碍患者一种有效的对症治疗选择(表3)。

在肌张力障碍患者中,BoNT主要通过对注入部位肌肉进行化学去神经支配,并导致其暂时性无力而起效。目前共发现7种具有不同免疫特性的毒素,且其都是通过干扰突触前神经终端乙酰胆碱的释放而发挥作用。A、B型肉毒毒素分别通过蛋白水解途径,裂解突触相关蛋白25(SNAP-25)和囊泡相关膜蛋白(VAMP)而起作用。Botox和Myobloc都在2000年获得美国FDA批准,用于颈部肌张力障碍的治疗。
最近几年,两个新品牌A型BoNT已经上市,分别是Dysport和Xeomin。Dysport已经FDA批准用于治疗颈部肌张力障碍;而Xeomin也被批准用于颈部肌张力障碍和眼睑痉挛的治疗。其他类型的肌张力障碍虽然没有专门的FDA许可,但已有研究表明,BoNT治疗口与下颌、喉、四肢和躯干部位的肌张力障碍,同样安全和有效。
肌张力障碍对BoNT反应差的原因有很多,其中包括剂量不足、肌肉选择不当、疾病相关的改变,以及某些患者对BoNT免疫抗药性的出现。但目前认为,现实生活中,对任何现代BoNT制剂和当前治疗指南推荐制剂产生中和抗体的患者较为罕见。
对一种品牌毒素出现耐药的患者,可以通过换用另一品牌而重新获益,但其对新制剂的反应,也会随着时间的过去而减小。调查这类药物作用过程的研究,大多局限于对Botox抗药肌张力障碍患者的Myobloc使用方面。
虽然不同品牌毒素制剂的结构和作用机制各不相同,但其长期疗效和副作用似乎没有显著差别。而且,目前尚缺乏这些制剂之间的比较。因此,毒素品牌的选择,可能会取决于其安全性和有效性以外的其他因素。
不同BoNT配方的配制和存储需求也有所不同,并可能在特定背景下,某一品牌的毒素可优于另一种。根据药物品牌和所在地区的不同,药物价格也会有所差异,并成为药品选择时的一个重要考虑因素。此外,当地的保险政策也会影响药物品种的选择。
在换用或使用不同配方的BoNT时,应特别注意其每种产品都有不同的特性,以确保不出现剂量错误、药物选择错误和不良事件。此外,面临对受教育程度越来越高的患者人群,有必要熟悉所有的BoNT制剂,以便应对患者的询问。
|
五大改变总结 ➤ 近日提出了修订的肌张力障碍定义和分类系统,这些新概念对有助于肌张力障碍诊断的临床特点进行了归类,并对正在快速增长,且有助于预示治疗发展的相关生物学机制进行了归纳整理。 ➤ 人们已越发认识到肌张力障碍的非运动症状对患者生活质量的影响,并已开始探索其病理生理意义。 ➤ 利用现代基因组测序方法,人们正在快速找出新的孤立性肌张力障碍基因,共同的分子通路也开始浮出水面。 ➤ 越来越多的证据已促成了以下新观点的出现,即应将肌张力障碍视为一种起因于脑区互联系统内部一个或多个受累区域功能障碍的神经网络性疾病。 ➤ 新型肉毒杆菌毒素制剂的出现,为肌张力障碍提供了更多安全、有效的治疗选项。 |
相关文章
评论

 我要跟帖
我要跟帖
.jpg)


