结合神经解剖与功能神经影像、小脑经颅磁刺激,PRRT2相关性阵发性运动障碍患者发生小脑功能障碍
发作性运动诱发性运动障碍(PKD)是一种短暂的、反复发作的肌张力障碍或其他不自主运动,通常由突然的自愿运动引起。PRRT2基因的单等位基因突变(hgnc: 30500)占PKD 家族性病例的90%,并且根据人群的不同,占散发性病例的一半。导致发作性运动障碍的原发性运动障碍的位置仍然是一个有争议的问题,根据病因可能会有所不同。纹状体和小脑在其发病机制中的各自作用可能是至关重要的。例如,与GTP环水
发作性运动诱发性运动障碍(PKD)是一种短暂的、反复发作的肌张力障碍或其他不自主运动,通常由突然的自愿运动引起。PRRT2基因的单等位基因突变(hgnc: 30500)占PKD 家族性病例的90%,并且根据人群的不同,占散发性病例的一半。导致发作性运动障碍的原发性运动障碍的位置仍然是一个有争议的问题,根据病因可能会有所不同。纹状体和小脑在其发病机制中的各自作用可能是至关重要的。例如,与GTP环水解酶Ⅰ缺乏相关的阵发性运动障碍可能是由于纹状体微回路中的多巴胺水平降低引起的,而且接受纹状体投射神经元输入的基底节主要输出GPI的损伤,也可能导致阵发性运动障碍。同样,双侧苍白球突变可以改善某些患者的运动障碍。相反,临床前期证据表明,小脑功能障碍在与ATP1a3突变相关的阵发性运动障碍中起关键作用。
PRRT2基因单等位基因突变的患者很少表现为阵发性共济失调,而双等位基因突变的患者可能有小脑萎缩,并且经常经历小脑共济失调的长时间发作。与这些观察一致,PRRT2的最高表达水平已经在小脑颗粒细胞中被发现。在这些颗粒细胞中特异性的PRRT2的抑制足以引起小鼠运动障碍。虽然以前对PKD患者的神经影像研究发现基底核、运动神经节和小脑脑前额叶外皮异常或功能障碍,但是这个还有待研究。近日,有研究人员在22例PRRT2相关PKD患者和22例健康对照组中,结合了全面的解剖和功能神经影像入路以及小脑的经颅磁刺激,试图研究了小脑功能障碍在PRRT2相关PKD发病机制中起关键作用的可能性。

研究共招募了22例发作性运动诱发性运动障碍合并PRRT2致病变异的连续右利手患者及其配对对照组。参与者接受了多模态神经影像检查。记录了解剖学和弥散加权MRI,以及静息状态功能MRI,在此期间测试了小脑内源性脑活动中假手术和重复经颅磁力刺激的后果。量化了: (i)灰质的结构完整性使用基于体素的形态测量; (ii)白质的结构完整性使用固定基础(iii)功能性小脑连接的强度和方向使用频谱动态因果建模。

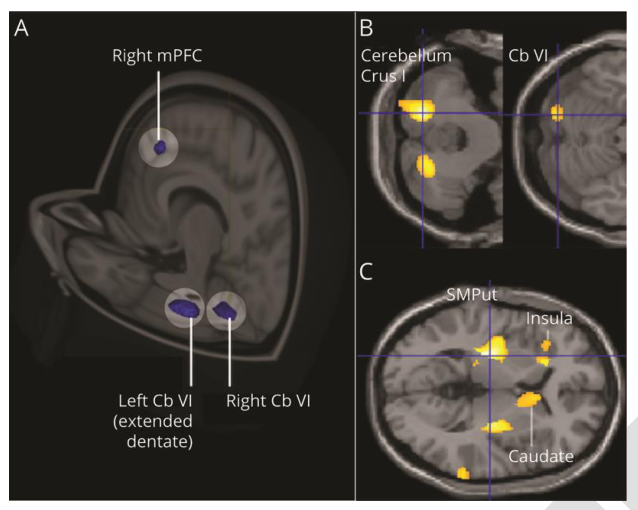



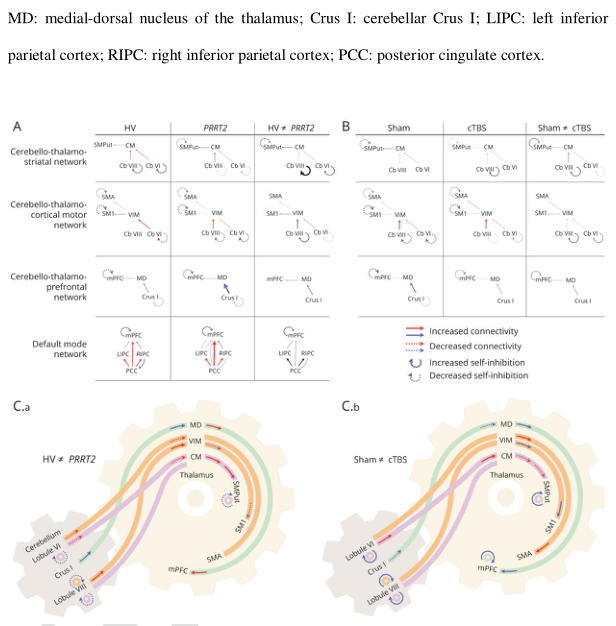
小脑 vi 叶和内侧脑前额叶外皮灰质体积减少;
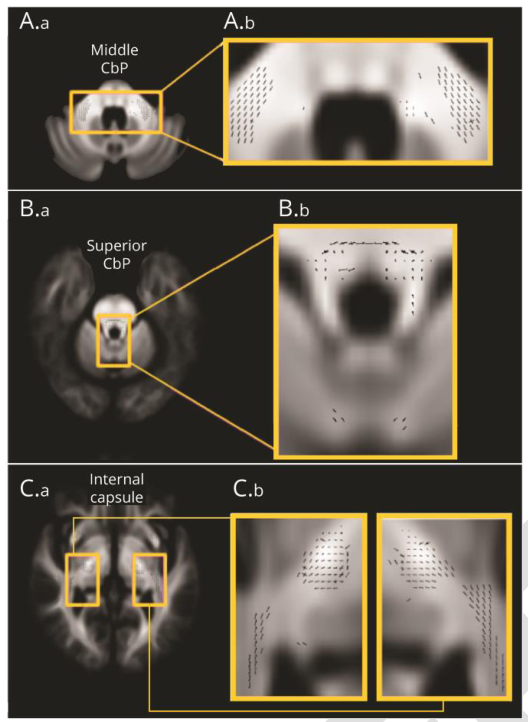
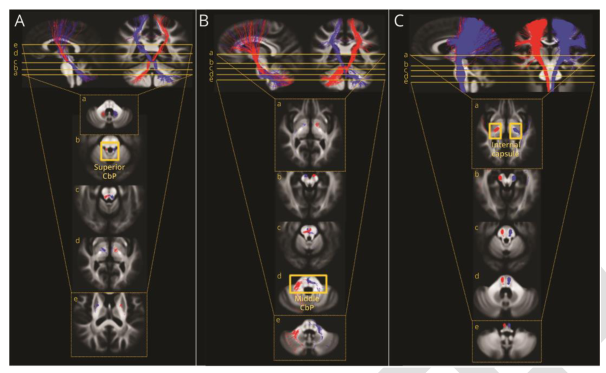
连接小脑、纹状体和皮质运动区的小脑束及小脑白质的显微结构改变;
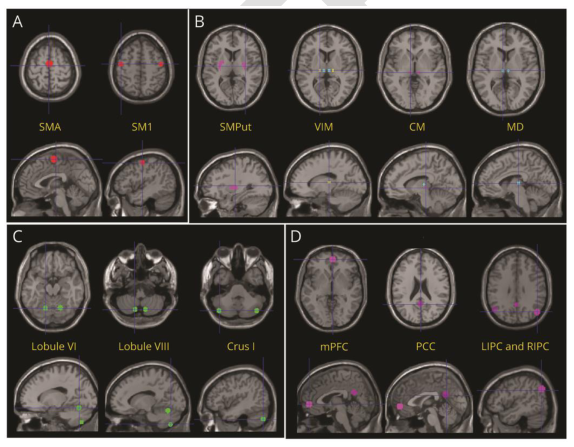
连接纹状体和皮质运动区的小脑运动通路功能障碍,以及小脑联合脑前额叶外皮与内侧运动区之间的异常通讯。小脑刺激调节了运动和联想小脑网络内的交流,并趋向于将这种交流恢复到健康对照组中观察到的水平。
PRRT2相关性运动障碍患者的运动小脑会聚结构改变和相关通路改变,小脑输出功能障碍通向小脑-丘脑-纹状体-皮质网络。假设PRRT2致病变异的病人,小脑输出异常是主要的功能障碍,导致纹状体调节障碍和阵发性运动障碍。更广泛地说,在某些疾病中,阵发性运动障碍的纹状体功能障碍可能是继发于丘脑继电器传递的异常小脑输出。
文献来源:Ekmen, Asya et al. “Cerebellum Dysfunction in Patients With PRRT2-Related Paroxysmal Dyskinesia.” Neurology, 10.1212/WNL.0000000000200060. 20 Jan. 2022, doi:10.1212/WNL.0000000000200060
相关文章
评论

 我要跟帖
我要跟帖




